|
||||||||
|
|
||||||||
|
Angelman sindromu
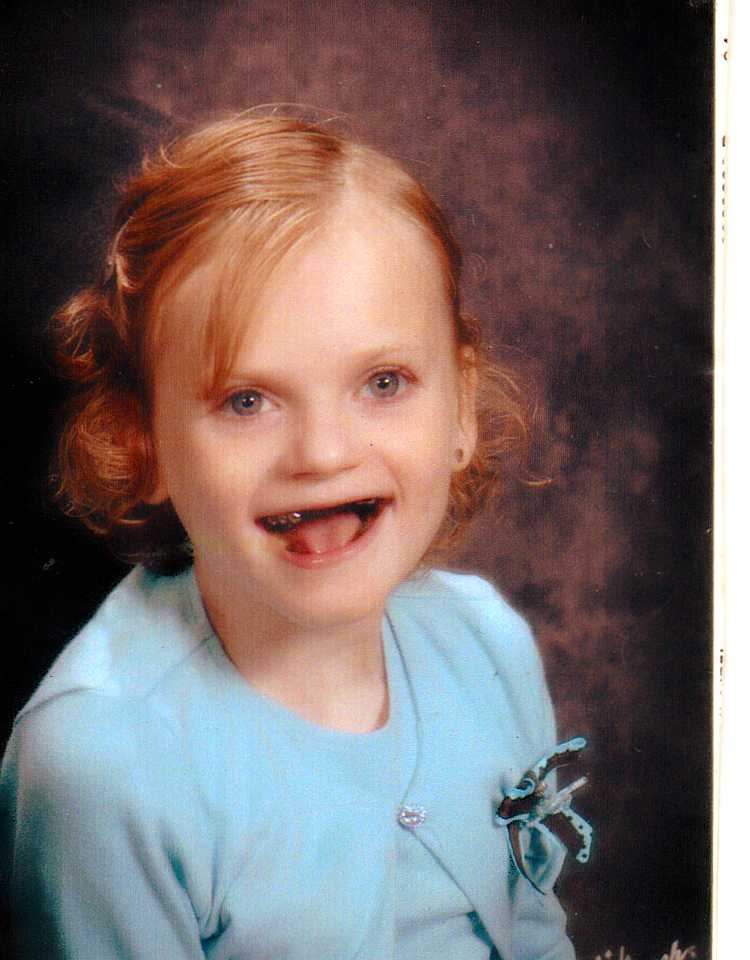
Angelman sindromu nadir rast gəlinən bir neyro-genetik xəstəlikdir.Irqlərdə görülmə tezliyi bilinməməklə birlikdə təxminən 15,000 ilə 30,000 təbii doğumdan bir olduğu qəbul edilir.Sindrom ilk dəfə 1965-ci ildə xəstəliyi təsvir edən İngilis həkim Harry Angelman'a ətf edilmişdir.Angelman Sindromu, ailə üzvlərindən yalnız birində müşahidə edilən;xromosom 15'də olan bir qrup gendəki problemdən qaynaqlandığı düşünülən bir xəstəlikdir.Xəstəliyin təməl tapıntıları zəka geriliyi,yerimə-koordinasiya pozuntusu,nitq pozuntusu,konvulsion və yersiz gülmə krizləridir.Hətta bu səbəblə xəstəlik bəzən "xoşbəxt kukla (happy puppet)" sindromu olaraq da adlanır. Lakin,xəstəliyin əlamətləri əksərən xəstə məktəb öncəsi yaşa gələnə qədər diqqət çəkmir və ya başqa xəstəliklərlə qarışdırılır.Angelman sindromlu körpələr normal hamiləlik və doğum hekayəsi,boy,çəki,başın ətraf ölçüləri baxımından normal fiziki xüsusiyyətlərlə doğulurlar.Ancaq ilk altı aydan sonra xəstədə inkişaf pozuntuları ortaya çıxır.İnkişaf gecikir ancaq degenerativ bir tapıntı və ya motor bacarıqların itirilməsi hallarına rast gəlinmir.İnkişaf geriliyinin xüsusilə müşahidə olunduğu ilk dövr 6 - 12 aylar arasıdır.Məhz burada ailənin diqqəti əhəmiyyət kəsb edir.Çünki bu uşaqlarda erkən diaqnoz ilə təmin edilən xüsusi təhsil dil və sosial adaptasiyanı asanlaşdırdığı kimi motor problemlərin həll edilməsində də böyük əhəmiyyət daşıyır.Angelman sindromlu fərdlərdə metabolik,hematologik və bütün biokimyəvi analizlər normaldır,ancaq Angelman sindromlu fərdin yaşı artdıqca danışma çətinliyi,ataksiya və inkişaf geriliyi,əlillik kimi hallar qeyd olunmağa başlayır. Angelman sindromlu fərdlər verbal ünsiyyət əvəzinə bədən dilini istifadə etməyə üstünlük verirlər və tipik asan xəbərdar olunan, tez gülümsəyən, maraq müddəti qısa və tez-tez əllərini çırpma tərzi hiper-motor fəaliyyət göstərən davranışlar nümayiş etdirirlər. Daha nadir olaraq bəzi Angelman sindromlu fərdlərdə xəstənin yaşı irəlilədikcə müəyyənləşən mikrosefaliya,çəplik,geniş ağız və seyrək dişlər kimi retarde bir üz ifadəsi ilə yanaşı ümumiyyətlə 3 yaşından sonra ortaya çıxan qıcolma tutmaları və EEQ'da pozuntular müşahidə olunur.Autizmin bir alt qrupu olaraq qiymətləndirilməsi də,autizmin bir çox davranış xüsusiyyətini özündə əks etdirir.Bəzən orta autizm diaqnozu də qoyula bilər.Autizmin bənzəri bir şəkildə,Angelman Sindromu olan fərdlər bu davranışları nümayiş etdirir:əl çırpma,çox az danışma və ya heç danışmama,diqqət əskikliyi,hiperaktivlik , qidalanma və adaptasiya problemləri və motor inkişafda geriləmə.Bu fərdlər həmçinin dişləmə və saç çəkmə kimi davranışlar da göstərə bilirlər.Autizmin əksinə bu insanlar daha çox sosial olaraq təyin olunur.Çox mehribandırlar və tez-tez qəhqəhə atırlar.Hərəkət və tarazlıq pozuntuları,ayaqların diskret dayanması,titrəməsi,Pals bədən duruşu,koordinasiya olmayan hərəkətlər müşahidə edilir.Dil,əmmə və yalama problemləri,dilin normadan böyük və çöldə olması,ağız suyu axması,həddindən artıq ağıza alma və çeynəmə davranışı problemləri müşahidə edilir və bu uşaqlar suya həddindən artıq maraq duyurlar. Angelman sindromunun molekulyar genetik mexanizmi qarışıq,bir o qədər də maraqlı bir xəstəlikdir. Molekulyar diaqnoz qoyulmuş pasiyentlərin əksəriyyətində 15. xromosomun uzun qolunun proksimal hissəsindən kiçik bir bölgə əksikdir ( 15q11 - q13 ). Buna görə də ümumiyyətlə bu sindrom "mikro - delesiya sindromları " qrupunda qəbul edilir.Ancaq maraqlı olan Angelman sindromu barəsində mikro- delesiyanın mütləq surətdə anadan gələn xromosomdan olmasıdır. Eyni mikro- delesiyanın atadan gələn 15. xromosomdan olması isə tamamilə başqa bir xəstəliyə,Prader - Willi sindromuna gətirib çıxarır. Bu faktın səbəbi xromosom bölgəsinin maternal və paternal kopyalarından fərqli şəkillərdə təzyiqlənmiş olmasındandır ( genetic imprinting ).Hər nə qədər Angelman sindromuna gətirib çıxaran gen və ya genlərin funksional olaraq hansıları olduğu mövzusunda işlər davam edir. Ancaq ilk dəfə bu xəstəliyin genetik siçan modelinin işarə etdiyi daha sonra edilən insan işlərində də diaqnoz qoyulmuş xəstələrin təxminən % 5indən mutasiyaya uğradığı göstərilən bu bölgədən bir gen ubiquitin ligaz olan UBE3A geni . Dahası siçan modeli xüsusilə hipokampus və beyincik bölgələrində xüsusilə maternal UBE3A geninin aktiv olduğunu və paternal genin demək olar ki,heç transkripsiya olmadığını göstərir. Angelman Sindromunun ən əhəmiyyətli xüsusiyyətlərindən biri də əldə edilən bacarıqların qalıcı olması; yaxşı bir təhsil ilə uyğunlaşma və bacarıqların inkişaf etdirilməsidir. Ancaq unudulmamalıdır ki,qəti bir müalicəsi yoxdur və həyat boyu davam edən bir xəstəlikdir. Nərmin Quliyeva |
BLOQMƏQALƏLƏR
LOGİN   |
|||||||
|
psixotest.az. Bütün hüquqlar qorunur 2012 ©
|
||||||||